MirAge is a fast analysing tool which is sensitive and highly precise. You can use any reference database build yourself.
It is capable of detecting Bacteria, Virus or any other DNA or RNA species. Even plain text analysis is possible to be used in i.e. plagiarism detection.
Configuration is intuitive, versitile and easy. High memory usage can be set if available, to speed up analysis.
MirAge can be used on your home PC, laptop or even a powerfull HPC (High Performance Computing) environment.
Especially for HPC usage, time estimation of the analysis is added so you can easily obtain an estimated required time to request a time window on your HPC environment.
Please do not hessitate to contact us at mirage@mirdesign.nl.
-
-


-
MirAge
DNA Taxonomic profiler -

-
MirAge
Download NCBI Reference database
To download and build a reusable NCBI Refseq database, use below unix commands to download and use the NCBI reference database.
- Unix
-
- 1. Retrieve complete NCBI refseq database
-
- wget https://ftp.ncbi.nlm.nih.gov/genomes/refseq/bacteria/assembly_summary.txt
- awk -F "\t" '$12=="Complete Genome" && $11=="latest" {print $20}' assembly_summary.txt > list
- awk 'BEGIN{FS=OFS="/";fs="genomic.fna.gz"}{fd=$0;asm=$10;f=asm"_"fs;print fd,f}' list > ftplist
- cat ftp.list | parallel -j 8 wget
- gunzip *_genomic.fna.gz
- cat *_genomic.fna >> ncbi.refseq.fasta
- rm *_genomic.fna
- Windows
-
- 1. Retrieve complete NCBI refseq database (Manual)
-
- Download https://ftp.ncbi.nlm.nih.gov/genomes/refseq/bacteria/assembly_summary.txt
- Open assembly_summary.txt in Excel
- Add a column at 21 next to the FTP path (column 20 (T))
-
Insert formula to all rows of your new column 21:
- = "curl " & T3 & MID(T3; FIND("!";SUBSTITUTE(T3;"/";"!";LEN(T3)-LEN(SUBSTITUTE(T3;"/";"")))); LEN(T3) - FIND("!";SUBSTITUTE(T3;"/";"!";LEN(T3)-LEN(SUBSTITUTE(T3;"/";""))))+1) & "_genomic.fna.gz --output " & ROW(A3) & ".fna.gz"
- Filter "Complete Genome" assembly_level (column 12 (L)) and "latest" version_status (column 11 (K)).
- Copy column 21 with the new URL's to a new plain file and save this as "download.bat"
- Execute download.bat
- Unpack all *.gz files:
-
- Use your already installed unpacker, or:
-
Download and install 7zip
-
"C:\Program Files (x86)\7-Zip\7z.exe" x *.gz -y
-
"C:\Program Files (x86)\7-Zip\7z.exe" x *.gz -y
- Put all *.fna into NCBI.refseq.fasta:
-
-
del *.gz
type *.fna > ncbi.refseq.fasta
del *.fna
-
del *.gz
- Get NCBI Taxonomic ID information
-
-
Now we have
ncbi.refseq.fasta containing the full complete genome database.
Per sequence, an accession ID is provided. To later be able to interper the results, taxonomy ID's are required. - 1. Retrieve Taxonomy data
-
- Unix
- wget https://ftp.ncbi.nlm.nih.gov/pub/taxonomy/accession2taxid/nucl_gb.accession2taxid.gz
- gunzip nucl_gb.accession2taxid.gz
- Windows
- curl https://ftp.ncbi.nlm.nih.gov/pub/taxonomy/accession2taxid/nucl_gb.accession2taxid.gz --output nucl_gb.accession2taxid.gz
- "C:\Program Files (x86)\7-Zip\7z.exe" x nucl_gb.accession2taxid.gz -y
- del nucl_gb.accession2taxid.gz
- 2. Use NCBI refseq database in MirAge
-
- ./mirage -reference ncbi.refseq.fasta -NCBITaxID nucl_gb.accession2taxid -query sample.fasta
- This will build a reusable ncbi.refseq.fasta.mhm database and analyse sample.fasta. The results provide TaxonomicID's and the provided header with Accession number and record name.
-
Now we have
ncbi.refseq.fasta containing the full complete genome database.
Configuration file
Override configuration.cfg
MirAge contains a configuration.cfg file which is used as primary setting source.
You can override each setting via the command prompt parameters directly.
As example, the configuration files contains progress_time_type with the default value 'remaining'. To override, provide the following parameter:
Settings explained
MirAge contains a configuration.cfg file which is used as primary setting source.
You can override each setting via the command prompt parameters directly.
As example, the configuration files contains progress_time_type with the default value 'remaining'. To override, provide the following parameter:
- mirage.exe --progress_time_type estimate
Settings explained
| Name | Possible values or (default) | Description | ||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| QuerySequencesPath | /home/username/data/sample1.fasta | The query file to be analysed Only 1 occurance allowed. | ||||||||||||||||||||
| ReferenceDatabasePath | /home/username/data/database.fasta /home/username/data/database.mhm | The reference file to be used Multiple occurances allowed, but only for MHM files. | ||||||||||||||||||||
| Hashmap_Load_Hashmap_Percentage | 1 to 100 (100) | Load a MHM database but randomly only load x% of the data. This is usefull when a quick scan of your sample is required. | ||||||||||||||||||||
| hashmap_load_hashmap_skip_on_failure |
0 1 |
End program when failing to load multiple hashmaps Skip hashmap when unable to load. |
||||||||||||||||||||
| Hashmap_Load_Reference_Memory_UsageMode |
|
|||||||||||||||||||||
| LogThreshold_HighSensitivity | 0, 0.1, 0.2, .. to 1 (0) | Threshold for high sensitivity log output. | ||||||||||||||||||||
| LogThreshold_HighPrecision | 0, 0.1, 0.2, .. to 1 (1) | Threshold for high precision log output. | ||||||||||||||||||||
| LogThreshold_Balanced | 0, 0.1, 0.2, .. to 1 (0.7) | Threshold for balanced log output. | ||||||||||||||||||||
| Hardware_BlockSize_Default_CPU | 1 or higher (5000) | Number of sequences to be analysed in 1 block. Higher requires more memory but can saturate CPU better. | ||||||||||||||||||||
| Hardware_maxCPUThreads | 1 or higher (75000) | Limit of number of threads used. | ||||||||||||||||||||
| Hardware_BlockSize_AutoIncrease_Enabled_Default |
1
0 |
Enable autoincrease to let MirAge try to optimise the block size in order to fully utilize the hardware. Disable autoincrease. | ||||||||||||||||||||
| Hardware_BlockSize_AutoIncrease_IncreasePercentage | 100 to 500 (130) | When autoincrease is enabled, how fast should it let the blocksize grow. | ||||||||||||||||||||
| UltraFastMode | Sensitive Fast |
Mode to analyse fast or sensitive. | ||||||||||||||||||||
| Output_Mode |
normal silent results resultsNoLog |
Full application log No screen output, only log files Show results on screen Show results on screen, without creating log files |
||||||||||||||||||||
| output_firstresults_only |
1
0 |
Only the references with the highest score are logged All scores and their related references are logged. Ordered by score. |
||||||||||||||||||||
| progress_time_type |
remaining estimate |
Show time remaining during analysis Show total time estimated during analysis |
||||||||||||||||||||
| considerthreshold | 1 and higher (32) | The threshold for accepting succesive regions. Lower is more sensitive and less precise. | ||||||||||||||||||||
| OutputPath | /home/user/output | The path where log and multinode files are stored. | ||||||||||||||||||||
| Screen_Resize (Windows only) |
0 1 |
Do not resize. Resize the command window |
||||||||||||||||||||
| Screen_COLS (Windows only) | 1 and higher (160) | Set window column size | ||||||||||||||||||||
| Screen_ROWS (Windows only) | 1 and higher (60) | Set window row size | ||||||||||||||||||||
| Hardware_Device_Queue_MaxSize (Experimental!) | 1 or higher (1) | Let the CPU do multiple prepare round while analysing a block | ||||||||||||||||||||
Publication
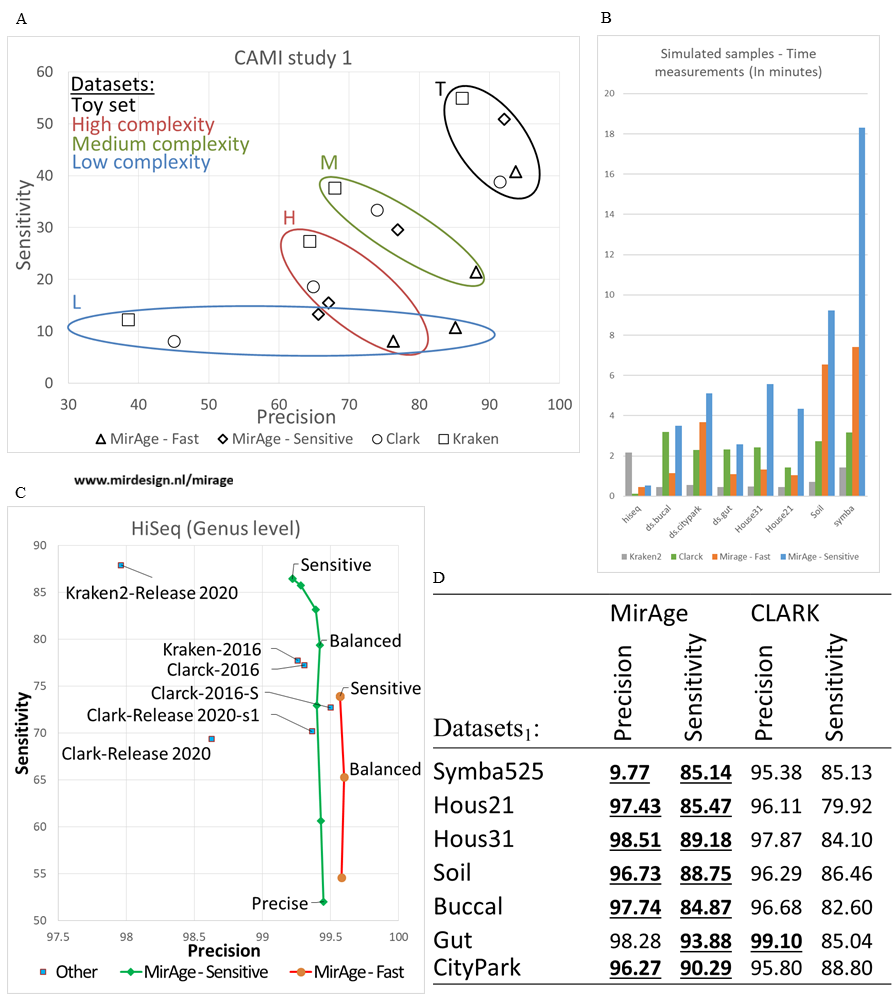
MirAge has been published on https://www.biorxiv.org/content/10.64898/2025.12.02.691787v1 (PDF)
Please citate MirAge as follow:
Please citate MirAge as follow:
M Pater, M D de Jong, MirAge, a novel Matrix Analyzer for fast DNA classification,
bioRxiv 2025.12.02.691787, doi: https://doi.org/10.64898/2025.12.02.691787